![]()
![]()
Resources on Isotopes
|
Chapter 16
Tracing Nitrogen Sources and Cycling in CatchmentsIsotope Tracers in Catchment Hydrology (1998), C. Kendall and J. J. McDonnell (Eds.). Elsevier Science B.V., Amsterdam. pp. 519-576. 16.1 Introduction
16.1 Introduction
Recent concern about the potential danger to water supplies posed by the use of agricultural chemicals has focused attention on the mobility of various solutes, especially nitrate and pesticides, in shallow hydrologic systems. Nitrate concentrations in public water supplies have risen above acceptable levels in many areas of the world, largely as a result of overuse of fertilizers and contamination by human and animal waste. The World Health Organization and the United States Environmental Protection Agency have set a limit of 10 mg/L nitrate (as N) for drinking water because high-nitrate water poses a health risk, especially for children,who can contract methemoglobinemia (blue-baby disease). High concentrations of nitrate in rivers and lakes can cause eutrophication, often followed by fish-kills due to oxygen depletion. Increased atmospheric loads of anthropogenic nitric and sulfuric acids have caused many sensitive, low-alkalinity streams in North America and Europe to become acidified. Still more streams that are not yet chronically acidic could undergo acidic episodes in response to large rain storms and/or spring snowmelt. These acidic "events" can seriously damage sensitive local ecosystems. Future climate changes may exacerbate the situation by affecting biogeochemical controls on the transport of water, nutrients, and other materials from land to freshwater ecosystems. The development of effective management practices to preserve water quality, and remediation plans for sites that are already polluted, requires identification of the actual sources and understanding of the processes affecting local nitrate concentrations. In particular, a better understanding of hydrologic flowpaths and solute sources is required to determine the potential impact of contaminants on water supplies. Determination of the relation between nitrate concentrations in groundwater and surface water and the quantity of nitrate introduced from a particular source is complicated by (1) the occurrence of multiple possible sources of nitrate in many areas, (2) the presence of overlapping point and non-point sources, and (3) the co-existence of several biogeochemical processes that alter nitrate and other chemical concentrations. In many circumstances, isotopes offer a direct means of source identification because different sources of nitrate often have isotopically distinct nitrogen and oxygen isotopic compositions. In addition, biological cycling of nitrogen often changes isotopic ratios in predictable and recognizable directions that can be reconstructed from the isotopic compositions. The main emphasis of the chapter will be on uses of isotopes in tracing sources and cycling of nitrogen in the water-component of forested catchments. However, the author has become convinced that watershed hydrologists not only have to look beyond hydrology to biogeochemistry, but must work towards an ecosystem or landscape approach to watershed processes. This kind of broad, interdisciplinary approach acknowledges that soil processes, deep groundwater reservoirs, agricultural and urban activities, and plant/animal functions all may have interacting roles in the watershed. Therefore, although the chapter focuses on dissolved nitrate in shallow waters, there will be brief mention of related subjects such as nutrient uptake studies in agricultural areas, large-scale tracer experiments, groundwater contamination studies, food-web investigations, and uses of compound-specific stable isotope techniques. This is not a chapter on environmental controls on nitrogen cycling in catchments; for good recent reviews of this topic see Cirmo and McDonnell (1997) and Stoddard (1994). And this is not a chapter on 15N-tracer approaches; for a good review of tracer methods, see Knowles and Blackburn (1993). 16.1.1 Fundamentals of nitrogen isotopes The whole-earth abundance of N is 0.03%, with 97.76% of the total N located in rocks, 2.01% in the atmosphere, and the remainder in the hydrosphere and biosphere (Hübner, 1986; from various sources). There are two stable isotopes of N: 14N and 15N. The wide range of oxidation numbers exhibited by nitrogen compounds, ranging from +5 (NO3-) to -3 (NH4+), results in a wide natural range of isotopic compositions. The average abundance of 15N in air is constant (Junk and Svec, 1958), with 15N/14N = 1/272. Nitrogen isotope ratios are generally reported in permil () relative to N2 in atmospheric air, using the standard definition of d (which is properly spelled and pronounced "delta" not "del"):
16.1.2 Methods Below is a brief description of how various N-bearing materials can be collected and analyzed for natural abundance isotopic composition, focused mainly on dissolved species. This is a rapidly evolving field with new methods being published every month. For up-to-date information, consult your library or the Web (see Chapter 2 for information about useful WEB sites and other analytical details). "Nitrogen Isotope Techniques" edited by Knowles and Blackburn (1993) provides a detailed, practical description of field and laboratory methods for N-related biological studies, mostly focusing on 15N tracer approaches, not the natural abundance applications that are the focus of this book. The chapter by Shearer and Kohl (1993) provides a description of natural abundance d15N applications, including a rebuttal to the skepticism of some ecologists about natural abundance applications of N isotopes. On the other hand, a recent review by Handley and Scrimgeour (1997) concluded that "the use of 15N is a powerful tool for obtaining insights through d15N pattern analysis, and for deriving new questions to be tested, but it is not a reliable tracer of nitrogen fluxes in soils or plants growing in soils." Although this chapter focuses on uses of N isotopes to understand water chemistry, isotopic compositions generally cannot be interpreted successfully in the absence of other chemical and hydrologic data. In particular, since redox reactions have such a profound impact on isotope fractionations, information on pH and dissolved oxygen concentrations are especially critical. A multi-isotope approach using the C, O, and S isotopic compositions of other organic and inorganic components will also aid interpretation of d15N variations in the ecosystem. How and where the samples are collected is as least as important as how and where they are analyzed. Adequate assessment of the temporal and spatial variability in potential endmembers is essential. In particular, sampling during fertilizer or manure application should be avoided because of rapid fractionations in the soil soon after application (i.e., ammonia volatilization and nitrification). Also, the nitrate-d15N values of materials applied at the surface (e.g., fertilizer, manure, treated waste) are best sampled beneath the application sites, after the N-bearing materials have been nitrified during downwards transport through the soil zone. Dissolved N-species
One method for collecting DIN from fresh waters for isotopic analysis is to use ion exchange resins (Hoering, 1957; Moore, 1977; Garten, 1992; Silva et al., in review; Chang et al., in review). Ammonium and nitrate are collected on cation and anion resins, respectively, in the field and the samples are sent to the lab for processing and analysis. The benefits of using exchange resins are: (1) elimination of the need to transport large volumes of water to the laboratory for processing, (2) elimination of the need for hazardous preservatives (e.g., HgCl2), and (3) ability to concentrate nitrogen from very dilute waters. Other anions and cations can successfully compete for exchange sites on resins, so such techniques are NOT suitable for saline or brackish waters; one must know something about water chemistry before using these techniques. Ammonium can be removed from the sample and converted to N2 gas for mass spectrometric analysis by (1) steam distillation of ammonium followed by oxidation with a hypobromite solution, and purification of N2 in a Cu/CuO furnace (Bremner, 1965; Bremner and Edwards, 1965); (2) distillation followed by collection of ammonium on an ammonium-specific zeolite (Velinsky et al., 1989) and sealed-tube combustion using Cu/CuO and CaO to produce pure N2 (Kendall and Grim, 1990); or (3) the so-called "micro-diffusion method" where ammonium is slowly diffused into an acid solution or onto acidified filter paper to produce ammonium sulfate, and combusted or reacted as above to form N2 (MacKown et al., 1987; Sigman et al., 1997; Downs et al., in press). Automated combustion of solid samples for C, N, and S (and now pyrolysis of samples for O and H) isotopes and elemental percentages on an elemental analyzer connected to a continuous-flow mass spectrometer has recently become a very popular method for analyzing more than 100 samples per day with analytical precisions of about 0.2 (except for d2H). This automated method can be applied to any of the methods below that produce solids (e.g., silver nitrate, ammonium sulfate, DON, etc), and to some liquids. Nitrate has conventionally been prepared by distilling off any existing ammonium, reducing nitrate to ammonium by a Kjeldahl reaction (Bremner, 1965; Bremner and Edwards, 1965), and then converting the ammonium to N2 using any of the above methods. Sigman et al. (1997) have adapted the micro-diffusion method for the analysis of seawater nitrate. A simple method, suitable for samples with negligible organic N, is to freeze-dry filtered samples, and then combust the resulting nitrate and other salts using Cu/CuO and CaO to produce pure N2 (Kendall and Grim, 1990). Using an ion exchange method to collect nitrate (Silva et al., in review), the nitrate is later eluted with HCl, neutralized with Ag2O, filtered to remove the AgCl precipitate, freeze-dried, and the remaining AgNO3 combusted to N2. This method is only suitable for fresh waters because high concentrations of anions in solution can interfere with nitrate absorption, causing a depletion of 15N in the sample. Samples can be archived, refrigerated on the column, for up to 2 years with minimal fractionation. Our group has used this method for over a thousand samples since 1991, and various modifications of it are being used by several other groups (e.g., Wassenaar, 1995; Aravena and Robertson, 1998; Harrington et al., 1998). Nitrate can also be analyzed for d18O. Amberger and Schmidt (1987) described a method whereby the sample was converted to KNO3, combusted with Hg(CN)2 to produce CO2 that was purified and analyzed for d18O. A modification of the method (Silva et al., in review) eliminates the need for hazardous materials such as mercuric cyanide by converting the sample to AgNO3 and combusting it with graphite to produce quantitative yields of CO2. Using this method to collect and process nitrate samples for d15N and d18O, analytical precisions for the laboratory standards put through the entire procedure are better than ± 0.1 for d15N, and ± 0.5 for d18O. For "real" samples, the error bars are about twice the size. An alternative laboratory combustion method (Revesz et al., 1997) that reacts samples in the form of KNO3 with catalyzed graphite gives excellent precision. Analysis on the automated pyrolysis system (Farquhar et al., 1997; Kornexl et al., in press) discussed above will probably soon become the preferred analytical method. Dissolved organic nitrogen (DON) is usually operationally defined as the N in dissolved organic matter (DOM) or dissolved organic carbon (DOC). Fractions of DOM rich in N include amino acids, proteins, and phenols. Because a wide variety of dissolved organic molecules contain nitrogen, exactly what is analyzed for DON-d15N depends primarily on how the water sample is processed after particulate N is removed. Methods for isolating the DOM for later combustion to N2 and analysis for d15N range from the very simple freeze-drying of the sample, to the addition of several possible methods prior to freeze-drying, including removal of DIN by ion chromatography, ultrafiltration to isolate a specific molecular weight fraction followed by ion chromatography (Bronk and Glibert, 1991), roto-evaporation followed by dialysis to remove DIN (Feuerstein et al., 1997), and passage through various resins to separate different fractions by their chemical properties (Aiken et al., 1979; Thurman and Malcolm, 1981). Other N-bearing species
Plants and animals are extremely simple to analyze for bulk isotopic composition, especially for d15N, d13C, and d34S. The samples are dried, ground, combusted, and analyzed -- generally using an automated system as described above. Gases can also be collected and analyzed for d15N (see Knowles and Blackburn, 1993), d18O, and other isotopes of interest. Gases are usually collected in a gas-tight syringes, bottles with septa-caps, or specially designed evacuated sample vessels. The samples may need to be combusted, and are then purified on a vacuum line or by passage through a gas chromatograph before analysis. An automated head-space sampler connected to the mass spectrometer can greatly reduce the manpower required for analysis. 16.2 The Nitrogen Cycle A schematic diagram of the nitrogen cycle in forest ecosystems is shown in Figure 16.1. Biologically-mediated reactions (e.g., assimilation, nitrification, and denitrification) strongly control nitrogen dynamics in the soil. These reactions commonly result in increases in the d15N of the substrate and decreases in the d15N of the product. Attempts to trace fixed N through the ecosystem are hampered by the complex fractionations caused by multiple cycles of mineralization, nitrification, immobilization, plant uptake, and denitrification within the soil (Lajtha and Schlesinger, 1986). Before describing some of these processes, we will briefly review some isotope geochemistry. 16.2.1 Isotopic fractionations Chemical, physical, and biological processes can be viewed as either reversible equilibrium reactions or irreversible unidirectional kinetic reactions; both kinds can have significant isotope fractionations. The processes are commonly modeled using Rayleigh equations (see below). This subject is discussed in detail in Chapter 2, and will be only briefly summarized here. The fractionation factor associated with the equilibrium exchange reaction A <--> B is defined as:
Kinetic fractionation factors can be defined as:
Readers of this book and articles dealing with isotope fractionations must be careful: both a and e values are defined in various ways by different authors. This chapter attempts to use the terms as "normally defined" for equilibrium systems (i.e., if s --> p, ap-s = Rp/Rs). However, these terms are commonly defined "in reverse" of normal usage in papers aimed at a biological audience to avoid using values of e < 0, or a values < 1. For example, the enrichment factor is sometimes defined in reverse (i.e., es-p), fractionation factors are commonly defined in reverse (i.e., a = Rp/Rs), many use the relation b = 1/a so that b > 1 (e.g., Figure 16.2), and some workers define a "discrimination factor" Ds/p = (as/p -1)*1000 where s/p denotes substrate relative to products. Good discussions of fractionations associated with biological processes include Hübner (1986) and Fogel and Cifuentes (1993). Readers should also keep in mind that the fractionation and enrichment factors determined in any particular study cannot readily be extrapolated to other studies because the degree of fractionation is strongly dependent on local conditions. Hence, the literature (including this chapter) contain a wide variety of ep-s values for any particular reaction. The Rayleigh equation describes the evolution of the isotopic composition of the residual reactant (substrate) during both kinetic and equilibrium processes (see Chapter 2). A commonly used formulation of the Rayleigh equation for systems with a constant fractionation factor is:
After all these equations, the reader might well have concluded that isotope fractionation is a very confusing concept. Not so! The idea is very simple: organisms preferentially use the light isotope (14N) over the heavy isotope (15N) so that almost anything created by the organism (the product) is isotopically lighter than the material not used (the reactant or substrate). For example, when microbes convert ammonium to nitrate (nitrification), the nitrate being formed is lighter (lower d15N value) than the ammonium being left behind. And as organisms use up the reactant, the d15N values of the product and left-over reactant change in predictable manner. As shown schematically in Figure 16.2, the d15N of the cumulative product N2 is always lighter than that of the residual reactant NO3. At the start of the reaction (reaction progress = 0), the d15N of the reactant is 0, and the d15N of the first bit of product is lighter than the reactant (by -5, -10, or -20, depending on which fractionation factor (b) is being considered). As the reaction progresses, the d15N of the reactant and the product both get heavier, and when all the reactant is used up (reaction progress = 1), the d15N of the cumulative product is equal to the starting composition of the reactant (d15N = 0) but the last bit of remaining reactant can have a very high d15N value. Therefore, the isotopic compositions of materials are highly dependent on the value of the fractionation factor and the size of the remaining reservoir of reactants. Many biological processes consist of a number of steps (e.g., nitrification: organic-N --> NH4+ --> NO2- --> NO3-). Each step has the potential for fractionation, and the overall fractionation for the reaction is highly dependent on environmental conditions including the number and type of intermediate steps, sizes of reservoirs (pools) of various compounds involved in the reactions (e.g., O2, NH4+), soil pH, species of the organism, etc. Hence, estimation of fractionations in natural systems is very complex. A useful "rule of thumb" is that most of the fractionation is caused by the so-called "rate- determining step" -- which is the slowest step. This step is commonly one involving a large pool of substrate where the amount of material actually used is small compared to the size of the reservoir. In contrast, a step that is not rate-determining generally involves a small pool of some compound that is rapidly converted from reactant to product; because the compound is converted to product as soon as it appears, there is no fractionation at this step. The isotopic compositions of reactant and product during a multi-step reaction where the net fractionation is controlled by a single rate-determining step can be successfully modeled with Rayleigh equations. 16.2.2 Processes affecting N isotopic compositions It is impossible to do justice to this complex topic in the space allowed. This section will highlight some of the major points of particular relevance to studying watersheds, with an emphasis on nitrate in waters. For an excellent review of nitrogen isotopes in soil-plant systems, see Högberg (1997). A monograph by Handley and Scrimgeour (1997), describing their many years of experience in studying nitrogen cycling in an abandoned field with isotopic techniques, provides a fascinating critique of d15N studies. Handley and Raven (1992) provides a general review of d15N applications in ecosystem studies. Griffiths (1998) contains many pertinent short articles on biological and ecological applications of isotopes. Fixation
Fixation of atmospheric N2 by blue-green and other bacteria (including those in N2-fixing root nodules, e.g., in legumes and alders) by the enzyme nitrogenase commonly produces organic materials with d15N values slightly less than 0. A compilation by Fogel and Cifuentes (1993) indicates measured fractionations (ep-s) ranging from -3 to +1. Because these values are generally lower than the values for organic materials produced by other mechanisms, low d15N values in organic matter are often cited as evidence for N2 fixation. The isotopic compositions of N-bearing materials produced by anthropogenic fixation (atmospheric gases produced during fossil fuel combustion, and artificial fertilizers produced from atmospheric gases) are discussed in Section 16.3.1. Common methods for quantifying biological fixation include the acetylene block technique and two isotopic methods: the 15N tracer dilution method (Warembourg, 1993; Watanabe and Wada, 1993) and the 15N natural abundance method (Shearer and Kohl, 1988). Perhaps the main benefit of the isotope methods is that the fixation estimate integrates over the entire life of the leaf analyzed instead of being an instantaneous rate. The natural abundance method has the further advantage that nothing has to be added and the disturbance to the ecosystem is limited to the sampling of leaves, soils, fertilizer, and air for isotopic measurement (Shearer and Kohl, 1988; 1993). A recent discussion of the topic (Högberg, 1997) notes that it is sometimes possible to obtain quantitative fixation-N data using the natural abundance method under carefully chosen conditions, and that sometimes this technique is the only practical method for assessing whether fixation is occurring. Assimilation
Measured values for apparent fractionations caused by assimilation by microorganisms in soils show a range of -1.6 to +1, with an average of -0.52 (compilation by Hübner, 1986). Fractionations by vascular plants show a range of -2.2 to +0.5 and an average of -0.25, relative to soil organic matter (Mariotti et al., 1980). N uptake by plants in soils causes only a small fractionation and hence, only slightly alters the isotopic composition of the residual fertilizer or soil organic matter. Fogel and Cifuentes (1993) present an elegant model for ammonium assimilation in aquatic algae that predicts total fractionations of -4, -14, or -27 depending on whether algae cells are nitrogen limited, enzyme limited, or diffusion limited, respectively. However, for the low pH values and low ammonium concentrations common to soils, the model predicts that availability of N is the limiting condition and the transport of ammonia across cell walls is probably rapid, resulting in a small (< -4) overall fractionation. A large range of fractionations (-27 to 0) has been measured in field and laboratory experiments for nitrate and ammonium assimilation by algae in aquatic environments (compiled by Fogel and Cifuentes, 1993). The much larger range of fractionations observed in aquatic environments vs. soil environments reflects the interplay of several possible kinetic and equilibrium isotope effects as a function of environmental conditions. In general, smaller fractionations are observed for higher growth rates and for lower NO3 and NH4 concentrations. The term dissimilation has been used to refer to N-metabolism because it can be viewed as the opposite of assimilation (Hübner, 1986). In metabolic reactions, the N compound is used as a supplier of energy by being either an electron donor (e.g., in redox reactions by nitrifying bacteria) or an electron acceptor (e.g., in the oxidation of organic compounds by denitrifiers). Mineralization
The reader should note that many other workers use the term mineralization for the overall production of nitrate from organic matter by several reaction steps. This usage results in literature that gives fractionations for mineralization that can range from -35 to 0, depending on which step is rate limiting (Delwiche and Steyn, 1970; Feigin et al., 1974; Letolle, 1980). The large fractionations are caused by the nitrification of ammonium, not the conversion of organic N to ammonium. In general, the d15N of soil ammonium is usually within a few permil of the composition of total organic N in the soil. Reviews of 15N tracer methods for determining mineralization and nitrification rates in soils and waters include: Mosier and Schimel, 1993; Powlson and Barraclough, 1993; and Glibert and Capone, 1993. Nitrification
oxidation by Nitrosomonas,
NH2OH + O2 ---> NO2- + [H] + H+ (Eq. 16.9) followed by oxidation by Nitrobacter,
The total fractionation associated with nitrification depends on which step is rate determining: one of the nitrification reactions listed above or the earlier production of ammonium from organic matter. Because the oxidation of nitrite to nitrate (Equation 16.10) is generally quantitative (rapid) in natural systems, this is generally not the rate determining step, and most of the N fractionation is probably caused by the slow oxidation of ammonium by Nitrosomonas. In soils, overall nitrification fractionations (b) have been estimated to range between 1.012 and 1.029 (Shearer and Kohl, 1986); i.e., the enrichment factors are -12 to -29 (d15NNO3 < d15NNH4). In general, the extent of fractionation is dependent on the size of the substrate pool (reservoir). In N-limited systems, the fractionations are minimal. Hence, the d15N of soil nitrate is usually within a few permil of the composition of total organic N in the soil. If there is a large amount of ammonium available (e.g., artificial fertilizer has been applied), nitrification is stimulated, and the oxidation of the fertilizer ammonium is the rate-determining step; this would likely cause a large fractionation. The d15N value of the first-formed nitrate is relatively low (Figure 16.3), but as the ammonium pool is used up, the nitrification rate decreases, oxidation of ammonium is no longer the rate-determining step, the overall nitrification fractionation decreases, and the d15N value of the total nitrate increases towards pre-fertilization values (Feigin et al., 1974). Therefore, one cannot accurately predict the d15N value of nitrate being leaked to surface water or groundwater from an agricultural field from simple measurement of the average d15N of ammonium fertilizers. The d15N of soil nitrate is commonly a few permil lighter (and sometimes heavier) than that of soil N because of fractionations associated with mineralization and/or nitrification. And even if the fertilizer applied were 100% synthetic KNO3 or guano, there would still be a possibility of post-depositional increases in d15N caused by denitrification as the nitrate was slowly transported to the sampling point. Increases in d15N of nitrate caused by denitrification are less likely in coarse-grained soils where waters percolate rapidly (and have higher concentrations of dissolved oxygen) than in finer-grained soils (Gormly and Spalding, 1979). Hence, the best way to assess the "effective" d15N value of the fertilizer or manure endmember is to collect samples from beneath the field where the materials are applied, avoiding sample collection soon after application since the fractionations are greatest then. Volatilization
Animal waste contains a wide variety of N-bearing compounds, both aqueous and solid, but most of the N is in the form of urea. The urea may be hydrolyzed to ammonia, and later oxidized (nitrified) to nitrate (Kreitler, 1975; Heaton, 1986):
Note that the above reaction consists of both reversible (equilibrium) reactions and irreversible (kinetic) reactions, but the overall reaction is unidirectional in that urea is irreversibly oxidized to nitrate. The hydrolysis of urea or ammonium fertilizer results in a temporary increase in pH, that favors the loss of ammonia gas by volatilization. The overall unidirectional reaction causes a preferential loss of ammonia depleted in 15N relative to the ammonium in solution (Figure 16.3). The loss of ammonia restores acidity and the remaining ammonium, now enriched in 15N, remains in solution. Much of the enriched ammonium is later nitrified to 15N-enriched nitrate (Figure 16.3). The degree of enrichment is determined by a variety of environmental factors that influence the rate of volatilization (e.g., soil pH, windspeed, moisture, temperature, etc). In a survey of fertilized soils in Texas, Kreitler (1975) attributed a 2-3 increase in d15N in underlying groundwater relative to the applied fertilizer to volatilization, and noted that losses of ammonia in alkaline soils can be very large and cause dramatic shifts in d15N. Sorption/desorption
Denitrification
Nitrate reduction by the heterotroph Pseudomonas denitrificans and the simultaneous respiration of CO2 from the oxidation of organic matter is the major cause of denitrification in soils:
There are several methods for determining the presence or extent of denitrification, including various enzyme-blockage methods (e.g., the acetylene blockage method) and 15N tracer methods (Mosier and Schimel, 1993). Natural abundance isotope methods include comparison of the increases in the (1) d15N of nitrate, (2) concentration and d15N of total N2, or (3) relative d15N and d18O of nitrate, with decreasing nitrate concentrations (see Section 16.5.2). The greenhouse gas N2O can be produced and released to the atmosphere by various mechanisms including denitrification in boggy soils and in aquatic systems near the sediment/water interface (e.g., Duff and Triska, 1990), and nitrification in soils. These two processes should be distinguishable isotopically because of differences in reaction mechanisms and kinetic fractionations. Further support for the source of the N2O can be gained by analyzing the d15N (and d18O) of other N-bearing compounds affected by the production of N2O. The N2O produced by nitrification is not likely to be metabolized in oxygenated waters, and will maintain its characteristic d15N and d18O values; in contrast, the d18O and d15N of N2O in anoxic conditions will increase because of consumption by denitrifiers (Yoshinari and Koike, 1994). 16.3 d15N Values of Nitrogen Sources and Reservoirs Most terrestrial materials have d15N compositions between -20 and +30. Although a recent compilation noted that the extreme d15N values for "natural" terrestrial substances reported thus far were -49 to +102 (Böhlke et al., 1993), these extreme values are the products of fairly unusual recycling of N; more typical ranges of major reservoirs are shown in Figure 16.4. The dominant source of nitrogen in most forested ecosystems is the atmosphere (d15N = 0); many plants fix nitrogen and organisms cycle this nitrogen into the soil. Other sources of nitrogen to watersheds include fertilizers produced from atmospheric nitrogen with compositions of 0 ± 3 and animal manure with nitrate d15N values generally in the range of +10 to +25; rock contributions of N to waters are almost always negligible. Note that fertilizer and animal waste have generally distinctive d15N values; however, the compositions of atmospheric and soil nitrate are not distinctive and overlap the compositions of fertilizer and animal waste. The d15N ranges of these N reservoirs at any single site are usually much less than shown on the figure. Two factors control the d15N values of any N-bearing compound in the subsurface (1) variations in the d15N values of inputs (sources) and outputs (sinks) of the compound in the subsurface, and (2) chemical, physical, and biological transformations of materials within the soil or groundwater that produce or remove the compound. Good reviews of the topic from different perspectives are given in Hübner (1986) and Högberg (1997). The sections below are intended to give the reader some general information about the isotopic compositions of various N sources or reservoirs, and how the N-cycling processes described in Section 16.2.2 affect these compositions. The discussion necessarily deals in generalizations derived at a limited number of sites, but the reader must not deduce from this that all ecosystems are alike, and the d15N values measured at one site cannot be blithely extrapolated to another. 16.3.1 Atmospheric sources Complex chemical reactions in the atmosphere result in a large range of d15N values of N-bearing gases and solutes depending on the compound involved, the season, meteorological conditions, ratio of NH4 to NO3 in the precipitation, types of anthropogenic inputs, proximity to pollution sources, distance from ocean, etc. (Hübner, 1986). Natural atmospheric sources of these gases and solutes include volatilization of ammonia from soils and animal waste (with fractionations as large as -40), nitrification and denitrification in soils and surface waters, and production in thunderstorms from atmospheric N2. Anthropogenic sources include chemical processing and combustion of fossil fuels in automobiles and power plants. The d15N values of atmospheric NO3 and NH4 are usually in the range of -15 to +15 (Figure 16.4). Extremely low d15N values for NO3 can be expected near chemical plants because of sorption of NOx gases (with high d15N values) in exhaust scrubbers (Hübner, 1986). There have been few comprehensive studies of d15N of precipitation, in part because of the difficulty of analyzing such dilute waters. Isotope shifts of several permil can occur between and within storms because of selective washout of N-bearing materials (Heaton, 1986), and the total range observed at any single location can be as large as 20. Studies in Germany (Freyer, 1978; 1991) and South Africa (Heaton, 1986; 1987) have found that d15N values of NO3 show a seasonal cycle of low d15N values in spring and summer rain and higher values in the winter. Freyer (1978) attributed this cycle to the release of depleted nitrogen oxides from soils (including nitrification of fertilizers) during the warm and moist growing season, and attributed the production of enriched NOx during colder seasons to the increased combustion of fossil fuels. Later work showed that variations in the d15N of NOx (i.e., its source) were not necessarily the main control on d15N of NO3 because of the large fractionation (~ +18) associated with the conversion of NO to NO2 in the atmosphere (Freyer et al., 1993). In general, the NO3 in rain appears to have a higher d15N value than the co-existing NH4 (Figure 16.4). For example, the average d15N values of NO3 and NH4 in Germany were -2.5 ±3.0 and -12.0 ±1.9, respectively; the lower NH4 values were explained by washout of d15N-depleted atmospheric NH3, and the higher NO3 values by washout of NO and NO2 (Freyer, 1978). Over a 1-year study at Walker Branch watershed (Tennessee, USA), the mean d15N values of NO3 and NH4 in precipitation (rain and throughfall) were +2.3 ±2.4 and -3.4 ±2.1, respectively (Garten, 1992); the lower NH4 values were again explained by washout of atmospheric NH3. Equilibrium exchange reaction of gaseous NO or NO2 with dissolved NO3 would result in 15N enrichment of the NO3. However, other studies have illustrated various complicated relations (Moore, 1977; Heaton, 1987), and there is considerable inter-storm and seasonal variability. The concentrations of N-bearing materials in precipitation are highly variable and often site-specific. Although precipitation in the eastern parts of the USA often contains subequal quantities of NH4 and NO3, NH4 is preferentially retained (utilized) in the tree canopy relative to atmospheric NO3 (Garten and Hanson, 1990), so that a larger proportion of the atmospheric nitrogen that reaches the soil surface is in the form of NO3. The mean d15N of red maple leaves in N-deficient ridges and slopes at the Walker Branch watershed is -3.2 ±1.2, similar to the composition of NH4 in bulk precipitation (Garten, 1992). This suggests that atmospheric NH4 might be a significant source of N for the trees, but further work is needed to verify this. Considerable attention has been given to nitrogen oxides (and sulfur oxides) in the atmosphere because of their contributions to acid rain. This is discussed in more detail in Chapter 22 (also see Heaton et al., 1997). Despite the complications of the atmospheric N cycle, isotope tracing of sources has been successful in some local studies. For example, there is some evidence that NOx emitted from coal combustion has a markedly different d15N value (+6 to +9) than NOx emitted from automobiles (-13 to -2), at least at the study area in South Africa (Heaton, 1990). The low values were attributed to kinetic fractionations in the formation of NO from atmospheric N2 and O2, and the high values to the d15N value of the coal (usually > 0) plus kinetic fractionations related to the breakdown of NO back to N2 and O2. Estimates for the d15N value of nitric acid vapor from anthropogenic sources range from -2.7 in Germany (Freyer, 1991), to +6.0 ±2.3 in Tennessee where 75-90% of the NO3 in dry deposition to an artificial tree was believed to be HNO3 vapor derived from coal combustion (Garten, 1995). The d15N of NO3 in dry deposition in Tennessee was about 6 heavier than in rain, close to the composition of soil nitrate. Hence, it is not surprising that throughfall, which contains dry deposition on the tree canopy, has a higher d15N value than rain. For example, the d15N value of NO3 in throughfall was found to be higher than in open-air rain, whereas the d15N of NH4 in throughfall had a variable composition relative to rain, in studies in Tennessee (Garten, 1992) and Yorkshire (UK) (Heaton et al., 1997). These findings suggests that throughfall-d15N is a better integrator of atmospheric N inputs to forested catchments than rain-d15N. Combined use of the d18O and d15N of nitrate (Section 16.4) may allow better separation of atmospheric and terrestrial nitrate sources (Amberger and Schmidt, 1987; Durka et al., 1994; Kendall et al., 1995b; in review; Böhlke et al., 1997), including the possible separation of different anthropogenic sources. Oxygen isotope ratios have proved useful for distinguishing N2O from nitrification and denitrification (Wahlen and Yoshinari, 1985). 16.3.2 Fertilizers Use of N-bearing fertilizers has a great impact on crop productivity, the d15N values of farmland plants, and on the N contents and d15N values of farmland soils. Overuse of fertilizers has resulted in high concentrations of nitrate, and significant changes in the d15N of the nitrate, in the surface waters and groundwaters issuing from farmland soils. Artificial (inorganic) fertilizers produced by the fixation of atmospheric N2 include the commonly-applied urea, ammonium nitrate, and potassium nitrate. These anthropogenic fertilizers have d15N values that are uniformly low reflecting their atmospheric source (Figure 16.4), generally in the range of -4 to +4; however, some fertilizer samples have shown a total range of -8 to +7 (see compilations by Hübner, 1986; Macko and Ostrom, 1994). Mean d15N values are (1) urea = +0.18 ± 1.27, (2) NH4 = -0.91 ±1.88, and (3) NO3 = +2.75 ± .76 (Hübner, 1986). Organic fertilizers (which include so-called "green" fertilizers such as cover crops and plant composts, and liquid and solid animal waste) generally have higher d15N values and a much wider range of compositions (generally +2 to +30) than inorganic fertilizers because of their more diverse origins. Note that the d15N of nitrate in fertilized soils may not be the same as the fertilizer. 16.3.3 Animal waste It has often been observed that animals (microbes to invertebrates) are slightly enriched in 15N relative to their diet, which is sometimes expressed as the isotope in-joke "you are what you eat plus 3" (or thereabouts). The increases in d15N in animal tissue and solid waste relative to diet are due mainly to the excretion of isotopically light N in urine or its equivalent (Wolterink et al., 1979). Animal waste products may be further enriched in 15N because of volatilization of 15N-depleted ammonia, and subsequent oxidation of much of the residual waste material may result in nitrate with a high d15N (Figure 16.4). By this process, animal waste with a typical d15N value of about +5 is converted to nitrate with d15N values generally in the range of +10 to +20 (Kreitler, 1975; 1979), and human and other animal waste become isotopically indistinguishable under most circumstances (an exception is Fogg et al., 1998). 16.3.4 Plants N-autotrophs can utilize a variety of materials from purely inorganic compounds (NH4, NO3, N2, NO2) to amino acids, and can have a wide range in d15N values depending on environmental conditions. However, most plants have d15N values in the range of -5 to +2 (Fry, 1991). Plants fixing N2 from the atmosphere have d15N values of about 0 to +2, close to the d15N value of atmospheric N2. N-heterotrophs (e.g., fungi) that utilize organically fixed N in the form of amino acids, have d15N values that are generally higher than soil N (Högberg, 1997). Recent investigations have concluded that there is negligible fractionation during plant uptake in most natural N-limited systems (Nadelhoffer and Fry, 1994; Högberg, 1997); nevertheless, tree tissues and litter have slightly lower d15N values than soil. Under higher nutrient conditions, preferential uptake of 15N by plants results in a few permil fractionation between plants and DIN. Whereas, in general, microorganisms and plants preferentially uptake ammonium, soil nitrate is preferentially assimilated by tree roots relative to soil ammonium (Nadelhoffer and Fry, 1988). Spatial variability in foliar d15N is commonly observed within forested catchments. A compilation of data for non-fixing trees by Garten (1993) shows as much as a 3-15 range in d15N values among the same species in small geographic areas. The large range reflects spatial variability in the relative amounts, d15N values, and bioavailability of atmospheric versus various soil sources of N; some examples of processes affecting variability are described below. The d15N values of non-fixing plants from a chronosequence in Hawaii (USA) increased substantially (-5.9 to +0.7) with age; soils showed a similar increase but were about 4 heavier (Vitousek et al., 1989). This increase with age was attributed to less reliance on 15N-depleted precipitation sources, higher rates of N cycling, more fixation and assimilation of N, and greater leaching losses in more mature soils. Foliar d15N values were higher on valley bottoms than on ridgetops in Tennessee (USA), reflecting the greater uptake of high-d15N soil DIN by plants in the valleys and greater uptake of low-d15N atmospheric ammonium on ridges where soil DIN is more limited (Garten, 1993). The 15N-enrichment of trees closer to the ocean relative to ones at higher elevations or at greater distances from the ocean perhaps reflects input of sea spray enriched in 15N (Virginia and Delwiche, 1982; Heaton, 1987). 16.3.5 Soils Nitrogen is recycled continuously between the atmosphere, soil, and the biosphere. The d15N of total soil N ranges from about -10 to +15, with most soils having d15N values in the range of +2 to 5. Cultivated soils had slightly lower d15N values (+0.65 ±2.6) than uncultivated soils (+2.73 ±3.4), according to a major soil survey by Broadbent et al. (1980). The d15N is affected by many factors including soil depth, vegetation, climate, cultural history, etc. Most of the N in soils is bound in forms not readily available to plants; hence, the d15N of total soil N is generally not a good approximation of the d15N of N available for plant growth. Soluble DIN (mainly NO3) constitutes about 1% of the N in soils, and hence is a very small pool whose d15N is much more sensitive to change than the larger organic pool. Turnover times of DIN in various soils are on the order of days (Davidson et al., 1990, 1992; Högberg, 1997). Because nitrate is more mobile in soils than ammonium, it is less likely to accumulate and, hence, readily leaches from soils. Although it has often been assumed that nitrate is the most abundant N solute in catchment waters, several recent studies have found that DON is actually the dominant N solute (Hedin et al., 1995; Gorham et al., 1998). The few DON-d15N values available for catchment waters are described in Section 16.7.2. There have been several investigations of the d15N values for soil nitrate from different environments (i.e., "natural" soils (tilled and untilled), soils fertilized with synthetic fertilizers or manure, soils contaminated with septic waste, etc). The data generated by a number of studies are summarized in Figure 16.4. In general, the soil nitrate produced from fertilizer (average d15N value = +4.7 ±5.4) and animal waste (average d15N = +14.0 ± 8.8) are isotopically distinguishable but they both overlap with the compositions of nitrate in precipitation and natural soils. However, given the large range of d15N values of the nitrate sources, the average values of sources from one site cannot be automatically applied to another; this is vividly illustrated by a recent compilation of nitrate d15N data (Fogg et al., 1998). Two factors, drainage and influence of litter, have a consistent and major influence on the d15N values of soil DIN (Shearer and Kohl, 1988). Nitrate in soils on lower slopes and near saline seeps has a higher d15N value than nitrate in well-drained soils (Karamanos et al., 1981), perhaps because the greater denitrification in more boggy areas results in 15N-enriched residual nitrate. The d15N values of nitrate in soils from valley bottoms at the Walker Branch watershed are higher than for soils from ridges and slopes, consistent with a theoretical model that explains the increase in the d15N of inorganic N in soil as a function of the higher relative rates of immobilization and nitrification in these bottom soils (Shearer et al., 1974). There is also greater uptake of atmospheric ammonium (which generally has a low d15N value) on the ridges because the limited availability of soil DIN there (Garten, 1993). It has recently been proposed that the release (drainage) of N from catchment soils can be explained by a flushing of the high-N upper layers of the soil during snowmelt or autumn storms, combined with a draining mechanism during snowmelt where recharge of the groundwater transports N from the upper soil layers into deeper flowpaths that contribute to baseflow throughout the year (Creed et al., 1996). Areas with abundant litter deposition (e.g., under trees and bushes) commonly have lower total d15N values than open areas (Shearer and Kohl, 1988; Nadelhoffer and Fry, 1988), presumably because losses of 14N to plant uptake during mineralization and nitrification in the open-area soil (i.e., natural soil processes) were not "offset" by the recycling of N from decaying litter. Discrimination against 15N during decomposition of litter (Melillo et al., 1989) results in the gradual 15N-enrichment of the residual organic material. Finer grained organic matter is generally enriched in 15N relative to coarser material (Tiessen et al., 1984). Well-drained soils typically show an increase in total soil-d15N with soil depth or with decreasing organic N content (Shearer et al., 1978; Shearer and Kohl, 1986). Nadelhoffer and Fry (1988) concluded that this increase in forests was due solely to fractionation during net mineralization, and not to differential preservation of components of litter with greater d155N values. Surficial soil organic matter d15N values are generally similar to or slightly greater than the values for plant litter; these values increase to about +8 ±2 at depths of 20-40 cm (Nadelhoffer and Fry, 1994). This increase in d15N with depth and age can be viewed as mainly the result of the metabolism of microbial heterotrophs that produce 15N-enriched biomass as a result of excreting 15N-depleted waste (Nadelhoffer and Fry, 1994). The loss of the bio-available, 15N-depleted ammonium to plant uptake, nitrification, and leaching coupled by recycling of the 15N-enriched biomass, will inevitably lead to increases in d15N of the total soil N. Accumulation of 15N-enriched, recalcitrant, mycorrhizal N with depth has also be suggested as an explanation for the increases in d15N (Gebauer and Dietrich, 1993). And there is some evidence that the d15N of DON also increases with depth (Sherry Schiff, pers. comm., 1998). Several investigators have reported that although nitrate d15N values usually increase with depth in surface soils, values can decrease below the rooting zone (50-500 cm) where N concentrations are low and N pools are mainly derived from leaching from above. Delwiche and Steyn (1970) noted that where there is a significant change in texture in the profile (e.g. a point where sand content is unusually high), there is a significant enrichment in 15N. But they could not demonstrate a consistent relationship between 15N content of N and soil particle size or total N content to the soil. Shearer et al. (1974) developed a theoretical model to explain the d15N of soil DIN as a function of the relative rates of N immobilization and nitrification. Although mineralization followed by nitrification and leaching are probably major causes of enrichments in total soils, other processes can also produce increases in d15N of nitrate with depth. For example, the inverse correlation of nitrate-d15N and nitrate concentration beneath agricultural fields (Gormly and Spalding, 1979; Böttcher et al., 1990) and in a forest (Koba et al., 1997) were attributed to increasing denitrification with depth. Seasonal changes in soil temperature may also affect the d15N of nitrate, resulting in higher values in the summer in unfertilized fields (Ostrom et al., 1998). In well-oxygenated vadose zones, there may be little or no change in the d15N of nitrate past the root zone, indicating little denitrification or other nitrogen cycling reactions during transport (Gormly and Spalding, 1979; Fogg et al., 1998) The complexity of the soil makes detailed studies of the different soil DIN pools difficult. For example, soil extractions using different soil:extractant ratios (Lindau and Spalding, 1984) and extractant chemical type (Burns and Kendall, in review; using DI, KCl and NH4Cl extractants) can cause more than a 6 range in nitrate d15N. Three possible explanations of these data have been proposed: (1) these values are artifacts caused by disturbance of the small, biologically active pools of N, (2) different pools of nitrate may have different d15N values, or (3) different pools are differentially available to flushing, perhaps because the nitrate pools are associated with different pore sizes or types of grain surfaces. Plants are integrators of the d15N of available N sources, and although there are complexities caused by storage effects, perhaps plants -- especially fine roots -- would provide the simplest and best estimate of the d15N value of available N in the soil (Högberg, 1997). 16.3.6 Groundwater In the last decade as nitrate concentrations in public supply wells have reached unacceptable levels in many parts of the world, it has become obvious that more attention needs to be paid to linkages between human activities on the surface and groundwater quality (Follett, 1989; Spalding and Exner, 1993). Nevertheless, groundwater is an often forgotten reservoir of nitrate in catchments. This is because many catchment hydrologists have not realized that there might be significant amounts of groundwater stored within the bedrock of the catchment, and many forest and agricultural biologists have paid little attention to processes below the root zone. All too often the bedrock is erroneously regarded as being impermeable and thus of little relevance to surface water hydrology, and to ecosystem processes on the landsurface and in streams. In fact, not only is groundwater the major source of water to streams in almost all catchments (see Chapters 1, 10-14, and 20-21), but because deep groundwater systems often extend beyond the catchment "boundaries" assigned from surface topography, these leakages can have a significant effect on catchment water and solute budgets. For more information on assessing the hydrologic properties of shallow and deep groundwater reservoirs, see Chapters 7 and 9. The main N-related processes in groundwater that affect catchment hydrology are probably denitrification, temporary storage, and transport to streams. How the nitrate-containing waters are transported to the stream has a dramatic effect on nitrate concentrations in streamwater (Böhlke and Denver, 1995). If waters containing high concentrations of nitrate that "escaped" below the rootzone are intercepted by tile drains or if these waters travel along deep flowpaths in oxidized aquifers before discharging vertically upwards directly beneath the streambed, the nitrate-rich waters could be discharged unchanged into the stream. On the other hand, if these groundwaters flow laterally through anoxic zones in adjacent riparian areas or through deeper unoxidized units where denitrification and other processes reduce the DIN contents, the groundwaters may be a significant sink for nitrate in catchments. This is illustrated by Figure 16.5 where the nitrate concentrations and d15N values in two adjacent streams are largely a function of the different flowpaths utilized. In this case, storage time was less important than the geochemistry of the geologic unit; the age ranges of waters discharging to both streams were similar. Several recent studies have found larger groundwater nitrate reservoirs in catchments than previously suspected (Kendall et al., 1995b; Williams et. al., 1997; Burns et al., 1998). 16.4 d18O Values of Nitrate Sources and Reservoirs The d18O of nitrate is a promising new tool for determining nitrate sources and reactions. Because much less is known about the d18O of various nitrate sources and the fractionations associated with different nitrogen cycling mechanisms, this discussion is separate from the discussion of d15N reservoirs (Section 16.3). Although several techniques have been developed since the 1980's for analysis of nitrate for d18O (Amberger and Schmidt, 1987; Kendall et al., 1996; Revesz et al., 1997; Silva et al., in review), there have been few applications of these methods, probably because all the methods are labor intensive and the first involves hazardous materials. Fundamentals: Oxygen has three stable isotopes: 16O, 17O, and 18O. Stable oxygen isotopic compositions are given in terms of 18O/16O ratios using the definition given above. The d18O values of nitrate are reported in relative to the standard V-SMOW. Figure 16.6 (see also color version) is a compilation of d18O and d15N values of nitrate. Surprisingly, there is almost an 80 range in d18O values, corresponding to a 30 range in d15N values. Most of the spread in d18O values is caused by precipitation samples, but there is also considerable variability in nitrate-d18O values in streams and soils. Although the oxygens in nitrate are thought to be derived from atmospheric O2 (about +23; Kroopnick and Craig, 1972) and environmental H2O (normal range: -30 to +5), the larger range of nitrate-d18O values indicates that oxygen isotopes in nitrate are fractionated from their source compositions during atmospheric processes. 16.4.1 Atmospheric nitrate There are limited data on the d18O of nitrate in atmospheric deposition, with almost nothing known about possible spatial or temporal variability, or their causes. The first published data on the d18O of nitrate in precipitation were from forests in Bavaria, in Germany (Voerkelius, 1990; Durka et al., 1994), and showed a relatively tight cluster of d18O values in the range of +55 to +75. A much larger range of values (+18 to +70) was observed for some 110 rain, throughfall, snow, and snowmelt samples from three forested USGS research sites in the USA (Loch Vale, CO; Catskills, NY; and Sleepers River, VT), with an average of +45 ±15 (Kendall et al., in review). A set of data (n = 62) from forests in north-western Germany (Muensterland, near Dortmund) show a range of about +23 to +58, with an average of about +36 ±9 (Bernhard Mayer, pers. comm. 1998), and a set from sites in east-central Canada show a range of about +28 to +51 (Sherry Schiff, pers. comm. 1998). Figure 16.7 shows all available nitrate-d18O values for precipitation (which includes values for rain, throughfall, snow, and snowmelt). Two different histograms are shown: (1) all data, and (2) only data from North America, most of which is from the three USGS sites mentioned above. The average nitrate-d18O value for the entire precipitation data set is +43.6 ± 14.6 (n=232). There are no statistically significant differences among different types of precipitation for the data set as a whole; however, there often are consistent differences for sample types at an individual site (e.g., nitrate-d18O values in snowmelt at Loch Vale are usually lower than the values in snow, perhaps because of infiltration by rain with lower nitrate-d18O values). Possible explanations for the large range in d18O values include fractionations associated with nitrate formation in thunderstorms, incomplete combustion of fossil fuels in power plants and vehicle exhaust, and photochemical reactions in the atmosphere. Some of these processes have been shown to fractionate nitrogen isotopes (Heaton, 1990). Given the large d15N range of nitrate and ammonium produced by different reactions and degrees of equilibration in the atmosphere (Heaton, 1987; Freyer, 1991), and the high d18O values reported for ozone and other nitrogen and carbon oxides in the atmosphere (Wahlen and Yoshinari, 1985; Krankowsky et al., 1995; Röckmann et al., 1998), it is likely that "natural" atmospheric nitrate has a wide range of d18O values too. Furthermore, since Heaton (1990) reported that the d15N of NOx from coal exhaust was about 10 heavier than NOx from automobile exhaust, it is possible that these different anthropogenic sources of atmospheric nitrate may also have characteristic d18O values. Heaton (1990) attributed the different d15N values to kinetic fractionations associated with the reversible reaction N2 + O2 <=> NOx + N; these reactions probably would cause similar kinetic fractionations of the O isotopes. Hence, the combined use O and N isotopes is likely to be useful for tracking different kinds of pollutants -- if not on a global scale than perhaps on a regional scale. What evidence is there that different natural and/or anthropogenic atmospheric nitrate sources might have different d18O values? The bimodal distribution of North American data in Figure 16.7 (and perhaps the non-normal distribution of the entire data set) show moderate evidence of at least 2 sources and/or processes affecting the compositions. The lower mode is centered around values of +22 to +28, and the higher mode (or modes) has values centering around +56 to +64. Prior to the first reported analyses from Bavaria, it had been speculated that "natural" atmospheric nitrate d18O values might be around +23, the d18O value of atmospheric O2. However, given the large d15N range of nitrate and ammonium produced by different reactions and degrees of equilibration in the atmosphere (Heaton, 1987; Freyer, 1991), and the high d18O values reported for ozone and other nitrogen oxides in the atmosphere, it is likely that "natural" atmospheric nitrate has a wide range of d18O values. More recently, some nitrate-rich salts from deserts in northern Chile and southern California (USA) that have d15N values near 0 and d18O values between +30 and +50 have been tentatively interpreted as evidence for long-term accumulation of atmospheric N deposition in hyper-arid environments (Böhlke et al., 1997). The best way to determine the pre-industrial atmospheric nitrate isotopic composition is by analysis of NO3 in ice cores. Until this is accomplished, all we have are speculations. However, it appears likely that the O in nitrate with d18O values close to that of atmospheric O2 is probably derived from the atmospheric O2, with the slight enrichments in 18O perhaps caused by kinetic fractionations during "back reaction" of NOx to O2. All the data reported for precipitation in Bavaria (+50 to +70) plot within the high-d18O mode. This part of Europe has high concentrations of nitrate in precipitation, many acid-rain damaged forests, and is downwind of highly industrialized parts of central Europe. Hence, one possible explanation for the high d18O values of nitrate in precipitation in Bavaria is that the values may reflect an anthropogenic pollution source, perhaps derived from fossil fuel burning. The samples from Muensterland, further from coal-burning centers of central Europe, have considerably lower d18O values. Although there is no obvious correlation between nitrate loading and the d18O values of nitrate among the three well-studied USA catchments, the lower values from sites in less-densely inhabited and industrialized Canada provide some support of this hypothesis. It should be noted that the bimodal distribution seen in North American samples is not apparent in the entire data set. In fact, there is even some evidence that there may be another "source" around +45 in the North American data set. The lack of a bimodal distribution in Europe may indicate that the sources are more interspersed or that there is better atmospheric mixing. It is possible that methodological problems are causing some of the variations seen in Figure 16.7. Analysis of nitrate for d18O is both time-consuming and analytically difficult, which is why there are so few data available. It should be noted that the Bavarian samples were all analyzed using the Amberger and Schmidt (1987) mercury cyanide method whereas all the other samples were analyzed using various modifications of the silver nitrate method of Silva et al. (in review). Furthermore, incomplete removal of DOC, a problem that has plagued users of both methods, can have a significant effect on the d18O of the nitrate, with some samples (e.g., throughfall and snowmelt) having probably higher concentrations than other forms of precipitation; contamination by DOC-oxygen probably results in mid-range d18O values. In our lab, "blanks" have d18O values in the range of +20 to +30 and we have noticed that small samples often have lower-than-expected d18O values, possibly because of contamination. Given the international interest in solving acid-rain problems, it is surprising that so little attention has been focused on the possibility of tracking different sources of atmospheric nitrate by its O and N isotopic composition. From the data presented above, it is not unreasonable to speculate that processes in coal-fired power plants may produce nitrate with high d15N and d18O values, car-combustion processes may produce nitrate with low d15N (and perhaps low d18O) values, and natural atmospheric processes appear to produce nitrate with low to intermediate d18O values and intermediate d15N values. More data are certainly needed to assess processes controlling the spatial and temporal ranges in isotopic composition. For example, because the d18O of the precipitation reflects changes in air-mass sources, there may be a correlation between water d18O and nitrate d18O values in precipitation samples. 16.4.2 Synthetic fertilizers and reagent Amberger and Schmidt (1987) analyzed a number of types of anthropogenic nitrates and determined that synthetic nitrate formed from atmospheric oxygen has a distinctive d18O value (+18 to +22). All three oxygens in this nitrate are derived from atmospheric O2 (+23), and hence the d18O values are similar to that of O2. 16.5 Tracing Sources and Cycling of Nitrate Under ideal circumstances, stable nitrogen isotopes offer a direct means of source identification because the two major sources of nitrate in many agricultural areas, fertilizer and manure, generally have isotopically distinct d15N values (Figure 16.4). Hence, under favorable conditions, the relative contributions of these two sources to groundwater or surface water can be estimated by simple mass balance. Soil-derived nitrate and fertilizer nitrate commonly have overlapping d15N values, preventing their separation using d15N alone (Figure 16.4). An early attempt to use natural d15N values to determine sources of nitrate in surface waters (Kohl et al., 1971) received a highly critical response (Hauck et al., 1972). This was partly because the use of the d15N values of fertilizer and animal waste to trace their relative contributions to groundwater is complicated by several reactions (e.g., ammonia volatilization, nitrification, denitrification, ion exchange, and plant uptake) taking place within the hydrologic system that can significantly modify the d15N values. Furthermore, mixing of point and non-point sources along shallow flowpaths makes determination of sources and extent of denitrification very difficult. Because of all these problems, attempts to use d15N for tracing the source and fate of nitrate in groundwaters and surface waters often have only limited success, despite the moderately good separation of d15N values (Figure 16.4). But it is interesting to note that the many subsequent isotopic studies of nitrate sources in groundwater did not elicit much controversy at all, perhaps because they considered in more detail the effects of denitrification and other d15N-altering processes. Many have speculated that analysis of the d18O of nitrate in conjunction with d15N would significantly improve our ability to trace nitrate sources and cycling. Figure 16.9 (see also color version) is a "simplified" version of Figure 16.6 (see also color version) and shows the normal range of d18O and d15N values for the dominant sources of nitrate. Nitrate derived from ammonium fertilizer, soil organic matter, and animal manure have overlapping d18O values; for these sources, d15N is a better discriminator. In contrast, nitrate derived from nitrate fertilizer or atmospheric sources are readily separable from microbial nitrate using d18O, even though the d15N values are overlapping. From the few dual-isotope studies of groundwater nitrate that have been conducted thus far (Böttcher et al., 1990; Aravena et al., 1993; Wassenaar, 1995), it is not yet clear how useful d18O will be in source characterization in groundwater. However, the dual isotope method has proved quite useful for source identification in some surface-water studies (Ging et al., 1996; Kendall et al., 1995b, 1996), as described in Sections 16.6.1 and 16.6.2. The following sections discuss various isotopic techniques for determining the relative contributions of different sources of nitrate (i.e., how to resolve mixing problems), and several different methods for recognizing and accounting for the impact of denitrification on isotopic compositions and water chemistry. Section 16.6 presents several case studies in more detail. But the reader must not be mislead into thinking that the successful solution of the mixing algebra insures that the source determinations are accurate. It is difficult to determine realistic isotopic compositions of proposed endmembers and assumptions of conservative mixing are always dubious when biologically labile materials are concerned. On a similar theme, Handley and Scrimgeour (1997) concluded their monograph on the application of d15N to ecosystem studies with several "words to the wise" about successful uses of d15N, including (1) don't overinterpret the data, and (2) be careful about attempting to apply "univariate isotope theory to multivariate field problems." 16.5.1 Mixing If nitrate in groundwater or surface water derives from the mixing of two different sources that are known to have distinctive d15N values, in the absence of any subsequent fractionations, the relative contributions of each can readily be calculated. Many articles have illustrated this point on d15N versus NO3 concentration plots, showing that mixtures must plot on a line between the two "endmember" compositions. However, such mixing lines are truly straight only when d15N values are plotted against 1/NO3 (see Chapter 2). On the standard d15N vs. NO3 plots, mixing lines are hyperbolic unless the NO3 contents of the endmembers are identical. An example of this is given in Figure 16.10a (from Mariotti et al., 1988) where two waters with nitrate concentrations of 0.2 and 10 mg/L mix together; note that the curvature of the mixing line is very slight for some mixing proportions. Unfortunately, life is rarely this simple. There are multiple potential sources of nitrate in various ecosystems, the sources rarely have constant compositions, and even if they did, the initial compositions may have been altered by various fractionating processes before or after mixing. Hence, estimates of relative contributions will often be only qualitative. In particular, denitrification can greatly complicate the interpretation of d15N values because the exponential increase in d15N of residual nitrate with decreasing NO3 content caused by denitrification can sometimes be confused with mixing of nitrate sources. For example, on Figure 16.10a, all three curves are almost linear for nitrate concentrations of 2 to 10. Hence, an incautious worker could try to interpret all three as mixing lines. However, as shown on Figure 16.10b, two of these curves are exponential relations resulting from denitrification, not mixing lines. Mixing of sources can sometimes be resolved by analysis of both the d18O and the d15N of nitrate (or other semi-conservative chemical tracers). This dual-isotope approach has three main potential benefits: (1) oxygen isotopic separation of some sources is greater than for nitrogen isotopes, allowing better source resolution by having two tracers, (2) some nitrate sources that are presently indistinguishable with d15N alone (e.g., fertilizer vs. soil nitrate, or atmospheric vs. soil nitrate) may be identified only when the d18O of nitrate is analyzed, and (3) oxygen isotopic compositions of nitrate vary systematically with nitrogen isotopic compositions during denitrification (as illustrated in Figure 16.9). Thus, in systems where the dominant sources of nitrate are isotopically distinctive, source contributions can -- in theory -- be determined despite significant denitrification. 16.5.2 Denitrification Denitrification is the process that poses most difficulties for simple applications of nitrate isotopes. Hence, for successful applications of nitrate isotopes for tracing sources, it is critical to (1) determine if denitrification has occurred, and, if so (2) determine what was the initial isotopic composition of the nitrate (which is a necessary prerequisite for later attempts to define sources). There are many methods for identifying and quantifying denitrification in groundwater; some of these are applicable to soils and aquatic systems too. The following discussion focuses on several of the most commonly applied field-geochemical methods for quantifying denitrification. Common biological methods for quantifying denitrification, conducted either in the laboratory or in the field -- including 15N tracer additions to chambers (Mosier and Schimel, 1993), acetylene inhibition chamber methods, acetylene inhibition soil- core methods, and denitrification enzyme assay techniques -- will not be discussed here. Geochemical signature
Enrichment in 15N
A large range of isotopic enrichment factors for denitrification (e = -40 to -5) have been calculated (see Hübner, 1986), determined in laboratory experiments (Delwiche and Steyn, 1970), measured in the soil (Mariotti et al., 1982), and observed in marine studies (Cline and Kaplan, 1975). However, Mariotti et al. (1988) noted that at many sites where denitrification in groundwater was identified by the above method, the e values showed a more narrow range of about -5 to -8. What causes this large range of observed enrichment factors? Mariotti et al. (1988) presented two hypotheses. One explanation is that the denitrification rate is the main control on the enrichment factor. Hence, denitrification is a first-order reaction where slow rates (caused by low temperatures or low quantities of electron donors) result in larger fractionations (Mariotti et al., 1982). Therefore, small e values near -5 suggest relatively rapid denitrification, and large fractionations, such as the -30 ±6 reported by Vogel et al. (1981) for denitrification in groundwater under the Kalahari desert, would indicate a slow denitrification rate. This model is consistent with the 14,000 years Vogel et al. (1981) estimated for the time required to account for present conditions in the Kalahari aquifer. An alternate explanation, elegantly presented by Mariotti et al. (1988), is that relatively impermeable aquifers may provide a sink for nitrate that effectively reduces the enrichment factor. For example, the porosity of chalk can exceed 40% of the total volume, yet 90% of the porosity is dead-end pores where the water is virtually immobile. In these pores, denitrification can proceed to completion, catalyzed by bacteria on the walls of the pores. Consequently, the nitrate concentrations within the pores are lower than in nearby flowpaths where waters travel more rapidly. This concentration gradient between the low-nitrate pores and the high-nitrate flowpaths, causes molecular diffusion of nitrate into the pores, which act as an effective sink for nitrate. Mariotti et al. (1988) further observe that the isotope effect associated with diffusion should be small or nonexistent, resulting in a smaller net enrichment factor. One consequence of this model is that a change in hydrologic conditions (e.g., an increase in pumping rate in the aquifer), should result in a significant decrease in denitrification potential. Mariotti et al. (1988) conclude that the use of nitrate-d15N to study denitrification processes is well suited to groundwater investigations, and is easier to apply than using the d15N of dissolved N2 because of (1) the relative ease of collection and preservation of nitrate samples compared to samples of N2 gas, (2) the complications associated with accurate determination of the fraction of N2 produced by denitrification, and (3) uncertainty whether there is a simple Rayleigh relation between the N2 produced by denitrification and reaction progress. Isotopes can also be used to study denitrification in soils (Delwiche and Steyn, 1970; Mariotti et al., 1981) and the hyporheic zone (McMahon and Böhlke, 1996). As discussed in section 16.3.5, the commonly observed increase in d15N in soils with decreasing nitrate concentration may, in part, be due to denitrification. A study by Koba et al. (1997) uses the relative changes in d15N, nitrate concentration, and water chemistry in soils to conclude that intermittent denitrification is occurring in anaerobic microsites of otherwise aerobic soils as the water table rises in response to storm events and pores become temporarily waterlogged. Excess N2
Several studies have evaluated the extent of denitrification in aquifers by analysis of the d15N of dissolved N2, and then calculation of the d15N of the excess N2 (Vogel et al., 1981; Wilson et al., 1990; Böhlke and Denver, 1995; McMahon and Böhlke, 1996; Böhlke et al., in review). The relative contributions of original atmospheric N2 and excess N2 can be estimated in several ways. If the atmospheric N2 content of recharge waters were only a function of temperature, the initial N2 content could be calculated (1) if the average recharge temperature were known, or (2) from the measurement of the noble gas composition of the sample (i.e., Xe, Ne, Ar, etc.) because these will behave conservatively in groundwater after recharge. Measurement of the N2/Ar ratio in groundwater is one way to estimate the excess N2 produced by denitrification. This ratio varies only slightly with temperature (37.3 at 5oC, to 38.3 at 20oC) in air-equilibrated water. However, solution of small air bubbles (with N2/Ar ratio of 83.5) during infiltration, because of increases in hydrostatic pressure as waters migrate downwards, causes groundwaters to have higher ratios than expected for the recharge temperature (Wilson et al., 1994). The amount of this entrained air can also be quantified by determining the "neon index" -- the ratio of the measured Ne content and the expected air-equilibrated Ne content at the derived recharge temperature (Wilson et al., 1994). Values greater than 1 indicate supersaturation with entrained air. A study of nitrate in a sandstone aquifer in England, concluded that N2/Ar ratios higher than 44 were evidence of N2 from denitrification (Wilson et al., 1994). Böhlke and Denver (1995) used the difference in the N2 contents of the suboxic-denitrified waters (NO3-free) and air-saturated water samples with the same Ar concentration as an upper limit for the amount of excess N2, as shown in Figure 16.11a. The upper limit of 135µM of excess N2 is equivalent to 270 µM reduced NO3. After adjustment for the small (0.7) isotopic enrichment caused by solution (Klots and Benson, 1963), the d15N of excess N2 can be calculated by simple mass balance. Figure 16.11b shows the estimated d15N values for the excess N2 in the samples plotted in Figure 16.11a range from +2 to +5. This range is indistinguishable from the range of d15N of NO3 in oxic groundwaters from the same location where no denitrification has occurred; complete denitrification should produce N2 with the same d15N as the initial NO3. The negative correlation between the d15N values of N2 and Ar/N2 ratio indicates that the dissolved N2 in the denitrified waters was a mixture of atmospheric N2 and N2 produced by denitrification. Enrichment in 18O and 15N of nitrate
Figure 16.9 can be used to illustrate a useful application of this characteristic enrichment in the 18O of nitrate for a situation where d15N values are ambiguous, based on a true situation. Local water managers were concerned that a public-water supply well downgradient from a heavily fertilized (KNO3) orchard had elevated NO3 contents. Although the managers were convinced that fertilizer was the source of the nitrate, they wanted "proof" and had a few samples analyzed for d15N. To their surprise, the d15N values ranged from +5 to +6, which did not provide support for their theory because this value is higher than the compositions expected for fertilizer (0 ±2). Were they wrong about the nitrate source? The measured values could indicate mixing with an additional source of nitrate (e.g., leaking septic tanks or local manure sources), that the fertilizer had a higher d15N value than expected (see Figure 16.4), or they could be caused by denitrification. Analysis of a few NO3 samples for d18O could have helped resolve this question because significant denitrification of fertilizer results in different d18O values than mixing of fertilizer and manure (Figure 16.9). Another powerful application of the dual isotope method takes advantage of the apparently constant ratios of 18O and 15N enrichment (Dd18O/Dd15N) factors during denitrification. If the ratio is constant, and the d15N and d18O compositions of the two potential sources of nitrate contributing to groundwater are known, are distinguishable, and do not show much scatter in composition, in theory the "original" relative contributions of these two sources to the nitrate in any sample of groundwater can be estimated from the d15N and d15O values of the nitrate (Figure 16.12). Furthermore, if the ratio is consistent over a wide range of field conditions, it should be possible to determine the source contributions in a two-source situation regardless of the effects of denitrification. Such estimations are not affected by the extent or timing of denitrification and mixing, or the spatial arrangement of the sources (i.e., point versus non-point sources). A more complete description of this model is given by Kendall et al. (1995c). 16.8 Summary The dominant use of isotopes in catchment research in the last few decades has been to trace sources of waters and solutes. Generally such data were evaluated with simple mixing models to determine how much was derived from either of two (sometimes three) constant-composition sources. The world does not seem this simple anymore. With the expansion of the field of isotope hydrology in the last decade, made possible by the development and increased availability of automated preparation and analysis systems for mass spectrometers, we have documented considerable heterogeneity in the isotopic compositions of various sources of waters and solutes, including nitrate. We are still grappling with how to deal with this heterogeneity in our hydrologic and geochemical models. A major challenge is to use the variability as signal, not noise, in our models; the isotopes and chemistry are providing very detailed information about sources and reactions in shallow systems, if only we can develop appropriate models to use the data. This integration of chemical and isotopic data with complex hydrologic models constitutes an important frontier of catchment research. |
|
|||||||||||
{kind=link}
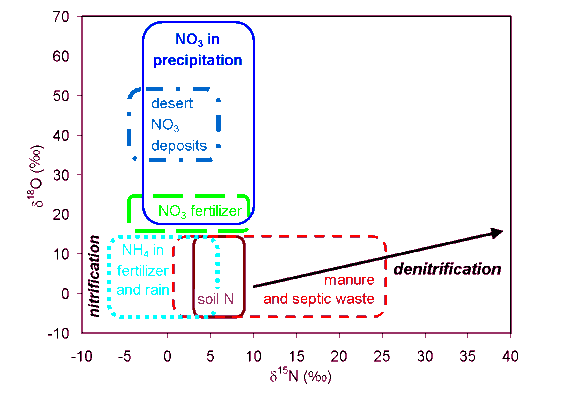{kind=link}
This page was last changed in April 2004.
To the USGS Home Page
To the Water Resources Information Home Page